
Highlights and Concerns in Thrombotic thrombocytopenic purpura TTP Treatment: From Pathophysiology to the Era of Caplacizumab
Raimondo De Cristofaro, Associate Professor of Internal Medicine, Faculty of Medicine and Surgery “A. Gemelli”, Università Cattolica del Sacro Cuore (Rome, Italy)
This article is part of the Garden Experts Insights series, featuring key perspectives from leading international experts who contribute to GARDEN educational initiatives and webinars.
In this edition, Prof. Raimondo De Cristofaro shares his insights on thrombotic thrombocytopenic purpura (TTP), discussing current challenges in disease management, emerging therapeutic strategies, and the evolving role of personalized approaches in improving patient outcomes. Drawing on both clinical experience and translational research, he highlights critical unmet needs and future directions in the treatment of this rare and potentially life-threatening thrombotic disorder.
Introduction
Haemophilia Thrombotic thrombocytopenic purpura (TTP) is a rare and potentially fatal blood disorder that, for much of the twentieth century, carried a mortality rate approaching 90%. The condition belongs to a broader family of syndromes known as thrombotic microangiopathies (TMAs), all of which share the hallmark features of microangiopathic hemolytic anemia and thrombocytopenia. TTP is distinct in its mechanism, its urgency, and above all, in the transformative therapeutic advances that have reshaped its management over the past three decades.
Today, with daily therapeutic plasma exchange (TPE) combined with corticosteroids, clinical remission is achieved in approximately 85% of patients.(1,2) The addition of rituximab as an immunosuppressive adjunct has further reduced relapse rates, and the approval of caplacizumab, a targeted anti-von Willebrand factor nanobody, has introduced a new era of mechanism-based acute therapy.(3) Yet despite these advances, TTP continues to present clinicians with genuine and pressing concerns: reaching the right diagnosis quickly when laboratory confirmation may take days, managing patients who fail to respond to standard treatment, preventing relapses in long-term follow-up, and recognizing the growing evidence that TTP is far more than an acute hematological emergency.
This review is prepared from the key insights of Professor De Cristofaro’s GARDEN webinar presentation, contextualized within current peer-reviewed evidence and the most recent ISTH guideline update published in 2025.(4)
Understanding Thrombotic Microangiopathy: A Framework for TTP
The term thrombotic microangiopathy describes a pathological state characterized by microangiopathic hemolytic anemia, thrombocytopenia, and thrombosis of the small vessels, producing damage across multiple organ systems.
Several conditions fall under this umbrella and distinguishing between them is not an academic exercise but a matter of therapeutic urgency.
The major causes of TMA include immune-mediated TTP (iTTP) and congenital TTP (cTTP), atypical hemolytic uremic syndrome (aHUS) driven by complement dysregulation, Shiga toxin-producing Escherichia coli associated hemolytic uremic syndrome (STEC-HUS), drug-induced TMA from agents such as calcineurin inhibitors, quinine, chemotherapy, interferon, mTOR inhibitors, VEGF or RTK inhibitors, and oral contraceptives, pregnancy-related TMA including HELLP syndrome, pre-eclampsia, eclampsia, and postpartum TMA, TMA associated with systemic diseases such as systemic lupus erythematosus (SLE), anti-phospholipid syndrome, scleroderma, malignant hypertension, glomerulonephritis, and malignancy, and metabolic forms related to MMACHC mutations affecting cobalamin metabolism.(5)
Pathophysiology of TTP: ADAMTS13 and the Von Willebrand Factor Axis
The Role of ADAMTS13
The molecular story of TTP centers on a single metalloprotease: ADAMTS13, a member of the ADAMTS (A Disintegrin and Metalloproteinase with ThromboSpondin motifs) family.This enzyme is a mono chain glycoprotein of approximately 190 kDa, made up of 1427 amino acids, synthesized predominantly in the liver and secreted into the bloodstream at a concentration of about 1 microgram per milli liter with a plasma half-life of roughly three days.(6) Its gene resides on chromosome 9q34 and encodes a multidomain protein comprising a pro-peptide, a catalytic metalloproteinase domain, a disintegrin-like domain, a first thrombospondin-1 repeat, a cysteine-rich domain, a spacer domain, seven additional thrombospondin-1 repeats, and two C-terminal CUB domains.
The physiological function of ADAMTS13 is to cleave ultra-large von Willebrand factor (UL-VWF) multimers that are released from endothelial Weibel-Palade bodies in response to injury or stress. Under the shear forces present in the microvasculature, these giant multimers unfold and expose the Tyr1605-Met1606 cleavage site within the VWF A2 domain. ADAMTS13 then cleaves this bond, reducing the multimers to smaller, less adhesive forms that circulate normally without spontaneously recruiting platelets.
When ADAMTS13 activity falls below 10% of normal, this cleavage does not occur. UL-VWF multimers persist at the endothelial surface, tethering platelets through the Glycoprotein Ib receptor on the platelet surface. Platelets adhere, roll, become activated, and are recruited in a cascade that culminates in the formation of platelet-rich microthrombi within capillaries and arterioles throughout the body. The resulting ischemia accounts for the neurological deficits, renal impairment, cardiac involvement, and hemolytic anemia that define TTP.(6,7)
Two subtypes of TTP arise from this mechanism. Immune-mediated TTP is the far more common form, caused by acquired inhibitory or depleting autoantibodies directed against ADAMTS13. Congenital TTP, also known as Upshaw-Schulman syndrome, is caused by biallelic loss-of-function mutations in the ADAMTS13 gene and represents a permanent, lifelong deficiency that typically presents in childhood or during pregnancy.
Measuring ADAMTS13 in the Laboratory
Quantification of ADAMTS13 activity in plasma is the cornerstone laboratory test for diagnosing TTP. The most widely adopted method uses fluorescence resonance energy transfer (FRET) technology, employing a modified fragment of the VWF A2 domain known as VWF73. In this assay, the substrate peptide carries donor and acceptor fluorophores flanking the cleavage site.
When ADAMTS13 cleaves the peptide, the energy transfer is disrupted and donor fluorescence is restored at 440 nm following 340 nm excitation. The rate of fluorescence change over time is directly proportional to ADAMTS13 activity.(8)
A critical practical limitation of current functional assays is that most are not sufficiently sensitive to precisely quantify activity levels below 10%. This is why severe ADAMTS13 deficiency is defined operationally as activity below 10%. The positive predictive value of this finding for TTP is high, because very few conditions other than sepsis, advanced malignancy, and disseminated intravascular coagulation produce a comparable degree of deficiency.(9,10) In contrast, both atypical and Shiga toxin-associated HUS are characterized by measurable ADAMTS13 activity above 10%, a distinction of fundamental therapeutic importance.
When ADAMTS13 activity confirms severe deficiency, the diagnostic workup is further refined according to subtype. In the acquired form, detection of anti-ADAMTS13 IgG inhibitor antibodies confirm the autoimmune etiology and carries important prognostic significance regarding relapse risk. In the congenital form, molecular genetic identification of ADAMTS13 mutations provides definitive confirmation and guides family screening.
Diagnosing TTP: Clinical Algorithms and the PLASMIC Score
One of the most important messages from Professor De Cristofaro’s presentation concerns the pragmatics of TTP diagnosis in real clinical settings. TTP is a medical emergency in which treatment must begin before confirmatory results are available.
The PLASMIC score, originally developed by Bendapudi and colleagues and subsequently validated in multiple independent cohorts including a large European study published in the British Journal of Hematology in 2019, assigns one point each for seven variables that can be obtained from routine hematological and biochemical tests available in virtually any hospital laboratory.(8,9) The score generates a stratified estimate of the probability that a patient with TMA has severe ADAMTS13 deficiency and therefore true TTP. It is presented in full in Table 1 below.
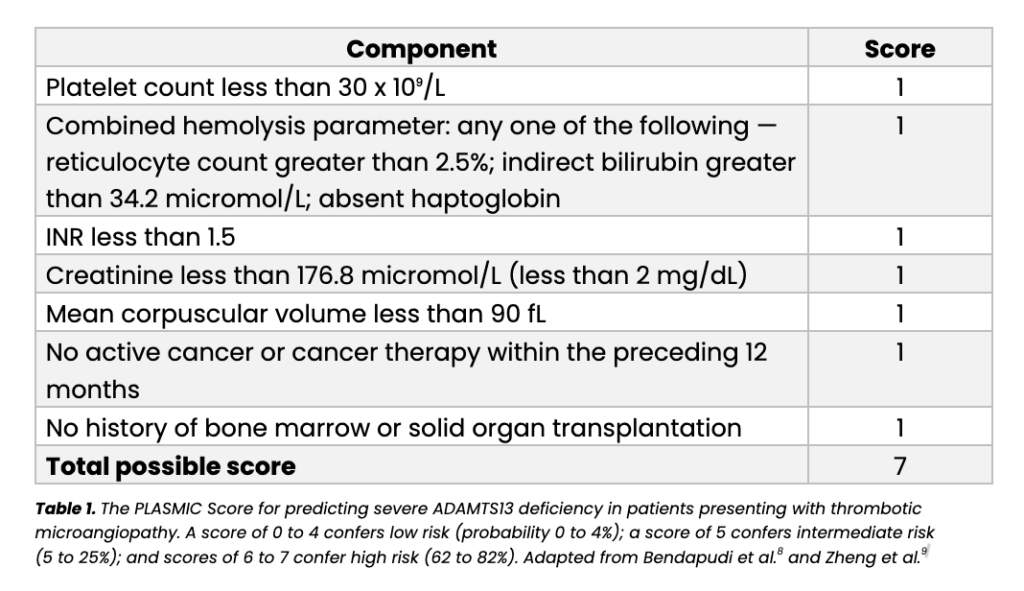 Beyond the PLASMIC score, the differential diagnosis between TTP, aHUS, and STEC-HUS follows a structured laboratory pathway. Any patient presenting with thrombocytopenia combined with microangiopathic hemolytic anemia confirmed by the presence of schistocytes, elevated lactate dehydrogenase, reduced haptoglobin, and falling hemoglobin, should have ADAMTS13 activity and Shiga toxin testing requested simultaneously. The three-way result determines the diagnostic category: ADAMTS13 activity at or below 10% points to TTP; ADAMTS13 activity above 10% with a positive Shiga toxin assay points to STEC-HUS; ADAMTS13 activity above 10% with a negative Shiga toxin assay raises the probability of aHUS and triggers complement pathway evaluation.
Beyond the PLASMIC score, the differential diagnosis between TTP, aHUS, and STEC-HUS follows a structured laboratory pathway. Any patient presenting with thrombocytopenia combined with microangiopathic hemolytic anemia confirmed by the presence of schistocytes, elevated lactate dehydrogenase, reduced haptoglobin, and falling hemoglobin, should have ADAMTS13 activity and Shiga toxin testing requested simultaneously. The three-way result determines the diagnostic category: ADAMTS13 activity at or below 10% points to TTP; ADAMTS13 activity above 10% with a positive Shiga toxin assay points to STEC-HUS; ADAMTS13 activity above 10% with a negative Shiga toxin assay raises the probability of aHUS and triggers complement pathway evaluation.
The Treatment Paradigm: From Plasma Exchange to Targeted Therapy – Therapeutic Plasma Exchange: The Historical Cornerstone
The publication of two landmark studies in the New England Journal of Medicine in 1991, the Canadian Apheresis Study Group trial by Rock and colleagues comparing plasma exchange with plasma infusion, and the clinical series by Bell and colleagues reporting outcomes in 108 patients, established daily therapeutic plasma exchange combined with corticosteroids as the standard of care for iTTP.(1,2) Before TPE, TTP was almost universally fatal. After its adoption, clinical and hematological remission rates reached approximately 85%.
TPE achieves its therapeutic effect through two complementary mechanisms. It removes the pathogenic anti-ADAMTS13 autoantibodies that are responsible for driving protease deficiency, and at the same time it replenishes functional ADAMTS13 activity from the donor plasma used to replace the patient’s own plasma. Corticosteroids add a further layer of immunosuppression to reduce ongoing autoantibody production.
Despite its efficacy, TPE-based treatment has well-documented limitations that create significant residual clinical burden. Exacerbations, defined as recurrent thrombocytopenia within 30 days of achieving a first remission, occur in approximately 50% of patients.(12) Refractoriness, defined as failure to achieve a sustained remission despite daily TPE and steroids, affects roughly 10% of patients and carries a meaningfully higher risk of death.(12) Procedural complications attributable to TPE itself including catheter-related infections, anaphylactoid reactions, citrate toxicity, and vascular access thrombosis occur in approximately 28% of cases.(13) These limitations created a clear medical need for adjunctive and alternative therapies.
Rituximab as a First-Line Adjunct
Rituximab, an anti-CD20 monoclonal antibody that depletes B lymphocytes and thereby suppresses autoantibody production, was first adopted for refractory and relapsed iTTP and has since been evaluated as a first-line adjunct to TPE. A phase 2 clinical trial by Scully and colleagues, published in Blood in 2011, enrolled 40 consecutive patients with newly diagnosed acquired TTP, all of whom received rituximab 375 mg per square meter weekly for four doses initiated within three days of admission, in combination with daily TPE and corticosteroids.3 The study demonstrated that early rituximab administration was associated with rapid recovery of ADAMTS13 activity, a significant reduction in relapse rates, and an extended disease-free interval compared with historical controls.
Current ISTH guidelines incorporate rituximab as a recommended first-line addition to TPE and steroids in newly diagnosed iTTP, particularly in patients with severe disease, high autoantibody titers, or prior relapse. It remains the preferred treatment for relapsed disease in patients who have previously responded.
Caplacizumab: Mechanism and Approval
Caplacizumab represents the first targeted therapy to receive regulatory approval specifically for acquired TTP, and it marks a genuine turning point in the acute management of this condition. The drug is a bivalent anti-VWF nanobody derived from the smallest functional fragment of heavy-chain-only antibodies naturally found in camelids such as llamas and alpacas, engineered to bind with high affinity to the A1 domain of VWF. By occupying this domain, caplacizumab physically blocks the interaction between VWF and Glycoprotein Ib on the platelet surface, rapidly interrupting the thrombotic cascade that underlies organ damage in TTP.(14)
The HERCULES Trial: Evidence Foundation
The pivotal phase 3 HERCULES trial, randomized, double-blind, and placebo-controlled, enrolled 145 patients with newly diagnosed or relapsed iTTP, defined as idiopathic TMA with a platelet count below 30 G/L and creatinine below 200 micromol/L.(14) Participants were allocated to caplacizumab 10 mg subcutaneously once daily or matching placebo, in addition to standard-of-care TPE and immunosuppression. The primary endpoint was time to platelet count normalization.
Caplacizumab significantly shortened the time to platelet count recovery, with a platelet normalization rate ratio of 1.55 (95% confidence interval 1.10 to 2.20; p less than 0.01).(14) The benefits extended across multiple clinically important secondary outcomes. The number of plasma exchange sessions required was reduced by 36% (5.7 sessions with caplacizumab versus 8.9 with placebo). The total volume of plasma transfused was reduced by 37% (21.1 liters versus 33.6 liters). The duration of ICU stay was reduced by 65% (3.4 days versus 9.7 days). The total length of hospitalization was reduced by 31% (9.9 days versus 14.4 days). These reductions represent a substantial decrease in the overall management burden for patients and healthcare systems alike.
The principal adverse effect of caplacizumab is an increased risk of bleeding, consistent with its mechanism of inhibiting platelet adhesion. In HERCULES, bleeding-related treatment-emergent adverse events occurred in 45.6% of caplacizumab-treated patients compared with 23.3% in the placebo group. The most frequently observed events were epistaxis (23.9% versus 1.4%) and gingival bleeding (11.3% versus 0%). Serious hemorrhagic events were uncommon and no fatal bleeding was attributed to caplacizumab.(14)
Real-World Evidence: The ROSCAPLI Study
Clinical trial results always raise the question of whether the observed benefits translate to routine practice. The ROSCAPLI study, a real-world multicenter Italian cohort published in the Journal of Clinical Medicine in 2024, addressed this directly.(15) In 38 patients with iTTP treated with caplacizumab outside of the controlled trial environment, the median time to platelet count normalisation was 2 days, with a mean of 2.69 days and a standard deviation of 1.26 days. These results closely mirror the HERCULES trial findings and confirm that the clinical benefits of caplacizumab are reproducible in everyday clinical settings.
Major Concerns: Relapse, Refractory Disease, and Long-Term Complications Relapse Risk and ADAMTS13 as a Prognostic Biomarker
Even after achieving hematological remission, TTP has a troubling propensity to recur. Cumulative relapse rates of 30 to 50% have been reported in follow-up studies extending over ten years.(11) A landmark survival analysis by Kremer Hovinga and colleagues published in Blood in 2010 demonstrated starkly that patients with persistently suppressed ADAMTS13 activity below 10% during clinical remission had dramatically worse relapse-free survival compared with those in whom activity recovered to 10% or above following treatment discontinuation.11 This finding established routine post-remission ADAMTS13 monitoring as an essential practice, identifying a high-risk subgroup who may benefit from pre-emptive rituximab therapy before clinical relapse occurs.
The timing of caplacizumab discontinuation is a related practical concern. The HERCULES trial continued caplacizumab for 30 days after the last plasma exchange session or until ADAMTS13 recovery was confirmed. Premature discontinuation before immunological remission is achieved carries a real risk of immunological exacerbation, a pattern observed in 28% of caplacizumab-treated patients in the trial who stopped the drug before ADAMTS13 activity had recovered. This has important implications for outpatient monitoring and for defining the criteria that should guide the decision to stop treatment.
Long-Term Complications: TTP as a Chronic Disease
Perhaps the most under-appreciated dimension of TTP is the burden of long-term complications that persist long after the acute episode has resolved. The evidence that has accumulated over the past decade makes it clear that TTP is not simply an acute hematological emergency that ends with platelet count normalization, it is a disease with chronic health consequences that demand sustained clinical attention.(16)
Depression and cognitive impairment are the most consistently documented sequelae. In a longitudinal follow-up study reported by James N. George and colleagues (Hematology, American Society of Hematology, 2019), 52 iTTP survivors were monitored from 2004 to 2014 using the Beck Depression Inventory-II (BDI-II) and the Repeatable Battery for Assessment of Neuropsychological Status (RBANS). Results were striking: 29% of patients exhibited severe depression at one or more assessment points, and only 41% showed minimal or no depression throughout the follow-up period. Cognitive testing revealed that 92% of assessed survivors demonstrated low average or below-average neuropsychological performance, with 20% scoring in the extremely low or borderline range, a prevalence of cognitive impairment far exceeding that expected in the age-matched general population.(16)
These neuropsychiatric sequelae almost certainly reflect the cumulative impact of cerebral microangiopathy and hypoxic-ischemic injury sustained during acute episodes, compounded by the psychological burden of living with a life-threatening relapsing condition. Beyond the neuropsychiatric domain, survivors face increased rates of systemic hypertension, autoimmune disorders including SLE, and renal impairment attributable to microvascular damage.
Emerging Therapeutic Approaches: Looking Beyond the Current Standard Recombinant ADAMTS13 for Congenital TTP
While this review has focused primarily on acquired iTTP, the management of congenital TTP deserves specific attention given a significant recent therapeutic development. Until the publication of the phase 3 cTTP trial in the New England Journal of Medicine in 2024, the only available treatment for Upshaw-Schulman syndrome was regular infusions of fresh frozen plasma or solvent-detergent-treated plasma to replace the absent protease — an approach that, while effective in preventing acute episodes, carries cumulative plasma-related complications and imposes a significant infusion burden on patients.(17)
The phase 3 trial of recombinant ADAMTS13 (rADAMTS13, BAX 930), conducted by Scully, Antun, Cataland, Coppo, and colleagues on behalf of the cTTP Phase 3 Study Investigators and published in the New England Journal of Medicine in 2024, demonstrated that subcutaneous administration of rADAMTS13 sustained ADAMTS13 activity and was associated with a meaningful reduction in acute TTP episodes compared with historical plasma-based treatment.(17) This represents the first disease-specific enzyme replacement therapy for cTTP and is expected to alter the management paradigm substantially for patients with this hereditary form of the disease.
Other Agents Under Investigation
N-acetylcysteine, which cleaves disulphide bonds within UL-VWF multimers and thereby reduces their thrombogenic potential, has been explored in observational and early prospective studies as an adjunct to standard therapy, though its role has not yet been established through definitive randomized controlled trial data. Development of recombinant ADAMTS13 for iTTP to supplement or replace endogenous deficient enzyme alongside immunosuppression, complement inhibitors, and next-generation anti-VWF nanobody derivatives represents the frontier of TTP therapeutics. These advances are the subject of ongoing translational and clinical research.
Current Guidelines: The 2025 ISTH Focused Update
The 2025 focused update of the 2020 ISTH guidelines for management of TTP, authored by Zheng, Al-Housni, Cataland, Coppo, and colleagues on behalf of the International Society on Thrombosis and Hemostasis, and published in the Journal of Thrombosis and Hemostasis in 2025, provides a structured algorithmic framework for integrating caplacizumab into contemporary clinical practice.(4)
For patients with a high pre-test probability of TTP defined as a PLASMIC score of 6 or above, or equivalent clinical judgement, when ADAMTS13 testing is not immediately available, the guidelines recommend initiating TPE plus steroids and caplacizumab simultaneously without delay. When ADAMTS13 testing is available or can realistically be returned within 24 to 48 hours and pre-test probability is low to intermediate, the recommended approach is to draw samples for ADAMTS13 testing, begin TPE and steroids, and defer the decision on caplacizumab pending results. If ADAMTS13 activity returns above 20%, caplacizumab should not be added and alternative diagnoses should be actively pursued. If ADAMTS13 activity is borderline at 10 to 20%, clinical judgement should determine next steps. If ADAMTS13 activity is below 10%, caplacizumab and rituximab should both be added to the treatment regimen.
The guidelines also reinforce rituximab as a recommended component of first-line therapy for all patients with confirmed iTTP, based on its established efficacy in reducing relapse rates and shortening the duration of detectable autoimmune activity. For relapsed disease, rituximab remains the cornerstone immunosuppressive agent, with escalation to other agents considered in cases of rituximab failure.
A Practical Approach to Modern TTP Management
Managing TTP well in 2025 requires a genuinely multidisciplinary team. Hematologists with expertise in thrombotic microangiopathies take the lead in diagnosis and treatment decisions. Intensivists manage hemodynamic support and oversee TPE logistics. Nephrologists are engaged when significant renal involvement is present. Neurologists assess and monitor cerebral complications. Clinical pharmacists support caplacizumab and rituximab dosing and safety surveillance. Specialist nurses, trained in apheresis catheter care and patient education, play a critical role in day-to-day management and in supporting patients through an experience that is profoundly frightening and physically demanding.
The initial workup in any patient presenting with suspected TMA should include a full blood count with differential, reticulocyte count, peripheral blood film examination for schistocytes, serum lactate dehydrogenase, haptoglobin, indirect bilirubin, creatinine, international normalized ratio, and direct antiglobulin test to exclude immune hemolytic anemia. ADAMTS13 activity and anti-ADAMTS13 antibody titer should be requested as a matter of urgency, and Shiga toxin testing should be sent simultaneously. While awaiting results, the PLASMIC score should be calculated from available data. A score of 6 or above should prompt immediate initiation of daily TPE and corticosteroids, with caplacizumab added according to guideline criteria.
Monitoring during the acute phase should include daily platelet counts, LDH, and hematocrit. ADAMTS13 activity should be retested at the point of remission and at regular intervals during follow-up at minimum monthly for the first three months and every three to six months thereafter. ADAMTS13 activity persistently below 10% during remission should prompt consideration of pre-emptive rituximab therapy before clinical relapse occurs.(11) Long-term follow-up should include structured neuropsychological assessment, blood pressure monitoring, autoimmune screening, and renal function evaluation.(16) TTP is a disease with chronic health implications that extend far beyond the normalization of the platelet count.
Conclusion
The history of TTP treatment is one of the more compelling stories in modern hematology. From a disease that was almost uniformly fatal, to one in which 85% of patients achieve remission with plasma exchange and steroids, to a condition now addressed with nanobody technology capable of directly blocking the molecular interaction responsible for acute microvascular injury, each step has been built on a deepening understanding of the underlying biology of ADAMTS13 and VWF.
The HERCULES trial has demonstrated unequivocally that caplacizumab accelerates platelet recovery and meaningfully reduces TPE requirements, ICU stays, and total hospitalisations.(14) Real-world data from ROSCAPLI confirm that these benefits translate outside the trial context.(15) The 2025 ISTH guidelines provide clinicians with a clear, risk-stratified decision framework.(4)
References
- Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med. 1991;325(6):393-397
- Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325(6):398-403
- Scully M, McDonald V, Cavenagh J, Hunt BJ, Longair I, Cohen H, et al. A phase 2 study of the safety and efficacy of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood. 2011;118(7):1746-1753
- Zheng XL, Al-Housni Z, Cataland SR, Coppo P, Geldziler B, Germini F, et al. 2025 focused update of the 2020 ISTH guidelines for management of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2025. doi:10.1016/j.jtha.2025.06.002
- Nester CM, Thomas CP. Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated? Hematology Am Soc Hematol Educ Program. 2012;2012:617-625
- Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488-494
- Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339(22):1578-1584
- Bendapudi PK, Hurwitz S, Fry A, Lindsley RC, Berlin DA, Niesvizky R, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4(4):e157-e164
- Zheng XL, Vesely SK, Cataland SR, Fortin PM, Hinds J, Jamula E, et al. British Journal of Haematology guidelines on the diagnosis and management of acquired thrombotic thrombocytopenic purpura. Br J Haematol. 2019;186(4):490-498
- Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585-1594
- Kremer Hovinga JA, Vesely SK, Terrell DR, Lammle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115(8):1500-1511
- Coppo P, Schwarzinger M, Buffet M, Wynckel A, Clabault K, Presne C, et al. Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies. PLoS One. 2010;5(4):e10208
- Howard MA, Williams LA, Terrell DR, Duvall D, Vesely SK, George JN. Complications of plasma exchange in patients treated for clinically suspected thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Transfusion. 2006;46(1):154-156
- Scully M, Cataland SR, Peyvandi F, Coppo P, Knobl P, Kremer Hovinga JA, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380(4):335-346
- De Cristofaro R, Iannaccaro P, Vitale R, et al. Real-world experience with caplacizumab in immune thrombotic thrombocytopenic purpura: the ROSCAPLI study. J Clin Med. 2024;13:6561
- George JN. Thrombotic thrombocytopenic purpura: a syndrome that keeps changing. Hematology Am Soc Hematol Educ Program. 2019;2019:336-343
- Scully M, Antun A, Cataland SR, Coppo P, Dossier C, Biebuyck N, et al. Recombinant ADAMTS13 in congenital thrombotic thrombocytopenic purpura. N Engl J Med. 2024;390(17):1584-1596

