
Extended Half-Life Therapies: What Real-World Practice Shows
Mariasanta Napolitano, MD, PhD
PROMISE Department, University of Palermo, Palermo, Italy
Haematology and Rare Disease Unit, “V. Cervello” Hospital, Palermo, Italy
This article opens a series of contributions featuring insights from speakers at our ECM webinars, now presented as editorial content.
We begin with a focus on extended half-life therapy, exploring what real-world clinical practice reveals and how current evidence is shaping treatment decisions.
Introduction
Haemophilia care has changed rapidly. Haemophilia A and B are congenital X-linked bleeding disorders caused by deficiency of factor 8 and IX respectively and severe disease which is defined by factor activity of less than 1% is commonly seen with spontaneous bleeding in early childhood (Chowdary et al., 2025). Today, the goal of care is no longer to simply find a way to stop a bleed when it occurs. The aim is to avoid bleeding, decrease the morbidity, preserve the joints and improve the daily life by the regular prophylaxis and comprehensive care (Chowdary et al., 2025; Srivastava et al., 2020). As Mariasanta Napolitano’s Webinar makes clear, treatment decisions no longer are limited to efficacy in well-controlled trials. Clinicians and patients also need to know whether a therapy is able to provide protection over time, minimize treatment burden in day-to-day practice, aid in adherence, and provide value in different healthcare settings. Those questions have taken on an even greater importance as prophylaxis options have expanded from standard factor replacement to extended half-life products, non-factor therapies and gene therapy (Carcao and Iorio, 2024; Rezende et al., 2024). Current guidance reflects this change. The 2024 ISTH clinical practice guideline made strong recommendations for prophylactic rather than episodic treatment in severe and moderately severe haemophilia and prophylaxis is recently being described as the standard of care for people with a severe bleeding phenotype (Rezende et al., 2024; Carcao and Iorio, 2024).
Burden of Haemophilia and the case of prophylaxis
The burden of haemophilia is accumulated. Repeated bleeding, particularly into joints, is not only associated with short-term symptoms but it is what drives chronic synovitis, haemophilic arthropathy, loss of function and reduced quality of life in the long term (Gualtierotti et al., 2021). Even subclinical haemarthrosis can contribute to joint remodelling, chronic pain and later disability, which is why avoiding the bleed is at the core of long-term care (Gualtierotti et al., 2021). Pain is now known to be a major clinical outcome in its own right. Recent evidence indicates that both acute and chronic pain impact across the life span people with haemophilia and can have physical, psychological, and social consequences that have a significant detrimental effect on health related quality of life (Benemei et al., 2024). Severe or repeated bleeding in the joints is important in particular as it may progress to chronic arthropathy with persistent articular pain and disability (Benemei et al., 2024).
This is the clinical background behind which prophylaxis was a turning point in haemophilia treatment. The WFH guidelines make regular prophylaxis the preferred strategy to prevent bleeding, morbidity and mortality and enhance quality of life and the 2024 ISTH guideline strongly favours prophylactic over episodic treatment for severe and moderately severe haemophilia (Srivastava et al., 2020; Rezende et al., 2024). Recent reviews have come to the same conclusion, with prophylaxis being the standard of care for people with a severe haemophilia phenotype (Carcao and Iorio, 2024). The evidence base is in favor of that shift. A 2025 Cochrane review concluded that clotting factor prophylaxis provides protection against overall and joint bleeds compared with episodic treatment, reinforcing the longstanding clinical move from reactive treatment to prevention (Razmpoosh et al, 2025). At the same time, the case for prophylaxis is expanding from severe disease. A review of non-severe haemophilia identified that individuals with high bleeding phenotypes may benefit from early prophylaxis to maintain joint health despite endogenous factor levels remaining above the severe range (Iorio et al., 2023).
Extended half life factor therapy in everyday practice
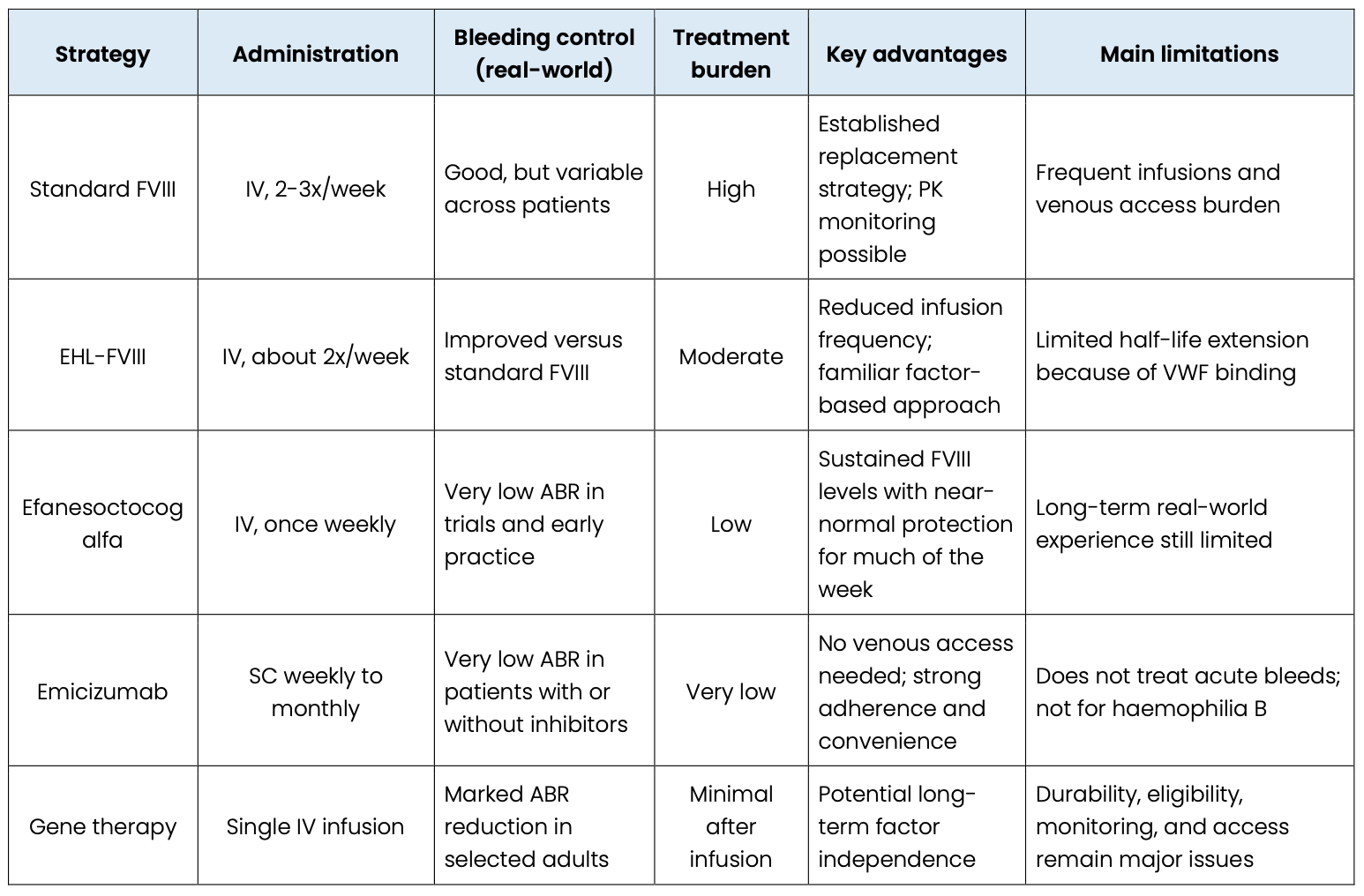
Extended half-life (EHL) factor products were developed to make prophylaxis more practical in the everyday haemophilia care. Their primary aim is to keep the levels of protective factors for longer, thereby requiring fewer intravenous infusions to be administered to the patient. This is important because not only do dosing schedules impact treatment burden, but so does a lot more. Frequent infusions can cause problems with school, work, travel, sport, venous access and long-term adherence. However, the benefit of EHL therapy is not the same as factors VIII and IX. Most EHL-FVIII products only increase half-life (1.3-1.7-fold), while EHL-FIX products typically result in a 4-6-fold increase in half-life (Chowdary, Khoo, et al., 2025). The primary reason is biological – FVIII is still limited by the binding to the von Willebrand factor that produces a ceiling effect. As a result, EHL therapy improved prophylaxis, both in haemophilia A and B, but the improvements have been much greater in haemophilia B (Lewandowska et al., 2025).
In the case of haemophilia A, real-life studies have demonstrated that EHL-FVIII products allow to reduce infusion burden and ensure good patient control of bleeds but still do not eliminate the need for regular IV therapy. In a real-world study in Padua of patients treated between 2018 and 2023, median infusion frequency in 2018 was 2.0 infusions per week with EHL-FVIII, while bleeding control across products was similar. Median half-lives were still modest at 16.5 hours for efmoroctocog alfa, 15.4 hours for Jivi and 19.8 hours for Esperoct (Zanon et al., 2025). This helps to support Napolitano’s message in the webinar that EHL-FVIII enhances convenience, but most patients still need treatment twice weekly or so rather than true long-interval prophylaxis.
There is a similar trend in the greater European literature. A 2023 systematic review of efmoroctocog alfa described low bleeding rates with median annualised bleeding rates generally being between 0.0 and 2.0, median injection frequency being between 1.8 and 2.4 per week and median weekly dose being between 60 and 105 IU/kg (Blatny et al, 2023). In the German Prevent study, the median ABR for patients treated with rFVIIIFc was 0.5 in 24 months and that switching from previous FVIII treatment was associated with a reduction of 56% in ABR (Bidlingmaier et al 2024). The A-SURE study also found that rFVIIIFc also had better bleed protection with fewer injections and lower factor use than standard half-life FVIII (Oldenburg et al., 2024). The picture is better in haemophilia B. Because the activity of FIX is not constrained by von Willebrand factor, the longer dosing intervals and more stable trough protection are achieved with EHL-FIX products. In a chart review of 138 patients aged 12 years and older in 2025, mean FIX consumption was 46.9 IU/kg/week with rIX-FP compared with 70.1 IU/kg/week with rFIXFc, while bleeding rates were low with all EHL products (Olivieri et al., 2025). A change from standard to EHL prophylaxis in children resulted in a longer median dosing interval (from 3.5 to 7 days), a greater mean trough level (from 4.3% to 15.3%) and lower factor use (from 66.0 to 37.0 IU/kg/week) (Dettoraki et al., 2025).
Unmet needs and the search for better protection
Extended half-life products improved prophylaxis, but they did not solve everything. The most significant limitation in haemophilia A is biological. Most previous EHL-FVIII products increase the half-life of the product only by approximately 1.3 to 1.7-fold because FVIII is still bound to von Willebrand factor, and this has a half-life ceiling of approximately 15 to 19 hours. Breakthrough bleeding is still possible, especially when trough levels are worsened, physical activity is increased or pharmacokinetics are variable from patient to patient (Lewandowska et al., 2025). In a prospective observational study of 157 adolescents and adults with severe haemophilia A, the median annualized bleeding rate on prophylaxis was 2.0, with only 35% of patients having zero bleeds, 18% having >5 bleeds in a year and mean number of routine prophylaxis injections being 2.2/week. The same study concluded that standard and extended half-life FVIII prophylaxis provided adequate haemostatic control in only around half of patients (Chowdary, Khoo, et al., 2025). A multinational non-interventional study in 33 countries, found continued burden as well, with treated-bleed ABRs of 4.7 in haemophilia A on prophylaxis and 10.3 in haemophilia A with inhibitors on prophylaxis, as well as continuing issues with joint health, physical activity and treatment burden (Wheeler et al., 2025).
Efanesoctocog alfa and the new generation of FVIII replacement
Efanesoctocog alfa is significant as all earlier generation EHL-FVIII products had had the major weakness of the von Willebrand ceiling (Yesim Dargaud et al., 2024). It is a B-domain deleted single chain F8 associated with the D’D3 domain of VWF and XTEN polypeptides and, therefore, can circulate independently of endogenous VWF. In review data, this design gives a much longer half-life of approximately 3 to 4 times longer than standard and previous EHL-FVIII products and supports normal to near normal levels of factor for most of the week (von Drygalski et al., 2023). The phase 3 XTEND-1 study revealed why this has got so much attention. Patients 12 years and above received 50 IU/kg once-weekly prophylaxis. In the main prophylaxis group of 133 patients, the median rate of annualized bleeding was 0, and the estimated mean ABR was 0.71. Mean ABR decreased from 2.96 on prestudy FVIII prophylaxis, to 0.69 on efanesoctocog alfa. Factor eight activity remained above 40 IU/dL for most of the week and was still 15 IU/dL at day 7 (von Drygalski et al., 2023). For clinical practice the message is quite clear: efanesoctocog alfa brings factor replacement close to the goal of good protection against haemorrhage with once-a-week treatment. That makes it a meaningful bridge between the traditional FVIII prophylaxis with the more recent expectation that treatment should also reduce burden, improve function and be compatible with real life.
Emicizumab in haemophilia care
Emicizumab has changed haemophilia A care as it addresses a problem that was of relevance in day-to-day life long before it was listed among trial endpoints: treatment burden (Jiménez-Yuste, 2024). A 2024 review which carried out a scoping review collating 97 publications on congenital haemophilia A found consistently low treated bleeds across both trial and real-world settings: calculated mean treated ABRs 0.7 to 1.3, median treated ABRs 0.0 to 1.4 and median proportion of people with zero treated bleeds was 66.7% (Young et al., 2024). The best, most powerful message from real-world research is that bleeding is falling, and that’s falling equally in inhibitor and non-inhibitor populations. In the PedNet registry analysis of 177 children, mean ABR in patients without inhibitors decreased from 2.41 before emicizumab to 1.11 after the start of emicizumab, where mean ABR decreased from 5.08 to 0.75 in the children with inhibitors; joint bleeding decreased significantly in both groups. That difference is important clinically because inhibitor patients have never had as many effective prophylactic options, and a larger treatment burden (van et al., 2024). The PedNet data therefore support what clinicians have seen in practice, which is emicizumab is valuable in non-inhibitor haemophilia A, but it is particularly transformative in inhibitor disease.
In the ATHN 7 haemophilia natural history study, 257 participants were on emicizumab at the cut-off date of December 2023; 24.5% of them had inhibitors at baseline, 84.4% had severe haemophilia A and 42.4% were under 12 years. Median treated-bleed ABR in participants with inhibitors 0.25; participants without inhibitors 0.51; indicating that the control of bleeds was maintained in both groups in routine practice rather than only in highly selected cohorts (Buckner et al., 2025). This is precisely the type of “real life” evidence that is relevant for clinical decision-making, because it reveals how a therapy works when it is used in an age and severity range and in different care settings. The usefulness of emicizumab is not limited to ABR in practice. In the webinar, Napolitano had an emphasis on convenience, adherence, and quality of life, and that is very consistent with the literature.
Safety in the real world has also been reassuring with one important caution. Across the 97-publication scoping review, most treatment-related adverse events were injection-site reaction (Young et al., 2024). In ATHN 7, the incidence of adverse events was 13 in 257 participants, and 33 injection-site reactions were reported in seven participants, and no thromboses or thrombotic microangiopathy were reported (Buckner et al., 2025). At the same time, writing in reality, real-world reviews stress that the breakthrough bleeds in patients on inhibitors should be treated with caution, as earlier thrombotic complications were associated with the use of high-dose activated prothrombin complex concentrate, rather than emicizumab alone (Kenet & Fujii, 2024).
Positioning EHL products and emicizumab in practice
In practice, the choice between extended half-life factor therapy and emicizumab is rarely about identifying one universally superior option. It is much more about selecting the approach that best fits the individual patient in front of you. EHL products still have the benefit of true factor replacement, being able to measure trough levels and pharmacokinetic tailoring which can be of use for active patients, perioperative planning and individualized dosing. Emicizumab offers something different: stable prophylaxis with subcutaneous administration, less dependence on venous access and strong real world bleed protection in haemophilia A with or without inhibitors. Recent guidelines state that there are no definite rules regarding the “best” first choice, or even an ideal time to switch and the decision to treat is still made on an individual basis and is heavily influenced by patient preference, bleeding phenotype, venous access, age and local access to care (Pratima Chowdary et al., 2024; Lewandowska et al., 2025).
 Real-world transition data bears that out. In the Czech National Haemophilia Programme registry, emicizumab switching was associated with a reduction of mean total ABR (3.44 to 0.46) and an increase in the proportion of patients with no reported bleeding (26.0 to 60.3% of patients) with a particularly strong benefit in younger patients and in those with inhibitors. At the same time, factors still play an important role because emicizumab is only prophylaxis, but does not treat acute bleeds, is not effective for haemophilia B and requires factors or bypassing agents for surgery and breakthrough bleeding (Zapotocka et al., 2025; Lewandowska et al., 2025).
Real-world transition data bears that out. In the Czech National Haemophilia Programme registry, emicizumab switching was associated with a reduction of mean total ABR (3.44 to 0.46) and an increase in the proportion of patients with no reported bleeding (26.0 to 60.3% of patients) with a particularly strong benefit in younger patients and in those with inhibitors. At the same time, factors still play an important role because emicizumab is only prophylaxis, but does not treat acute bleeds, is not effective for haemophilia B and requires factors or bypassing agents for surgery and breakthrough bleeding (Zapotocka et al., 2025; Lewandowska et al., 2025).
Gene therapy and the initial results in real life
Gene therapy is the most ambitious change in modern haemophilia care as it seeks to minimize or even eliminate the need for long term prophylaxis following a single infusion. In routine practice, however, the term “early real-world outcomes” needs to be used carefully. Post-approval real world datasets are, however, limited, so the best evidence to date remains from mature phase 3 follow-up and early implementation programmes rather than large post-marketing registries. Even so, the direction is clear – gene therapy has the potential to bring major reductions in bleeding and factor use in selected patients as adults, particularly in haemophilia B, but durability, eligibility, liver monitoring and long-term follow-up are key issues (Samelson-Jones et al., 2024; Chowdary et al., 2024). For haemophilia A, a gene therapy using a recombinant adeno-associated virus (AAV) known as valoctocogene roxaparvovec was the first licensed AAV gene therapy. In the phase 3, GENEr8-1 study, 134 men with severe haemophilia A were infused with the gene therapy once and at week 104, the mean annualized treated bleeding rate had decreased 84.5% from baseline. No new safety signals or treatment-related serious adverse events were observed at two years. That is a clinically important result because it shows that gene therapy can move some patients from the severe form of the disease towards a milder bleeding pattern. At the same time, a major note of caution with haemophilia A gene therapy is still durability. Later reviews of Roctavian highlight year on year decline in FVIII expression as the important unanswered question about longer-term clinical use (Mahlangu et al., 2023; Samelson-Jones et al., 2024). The case of haemophilia B has so far been more stable. In the HOPE-B phase 3 study of etranacogene dezaparvovec, mean adjusted ABR decreased from 4.18 during the lead-in period, to 1.51 in months 7 to 24 after gene therapy and factor IX-treated bleeds decreased from 3.65 to 0.99. By 24 months 52 of 54 participants or 96% remained free from FIX prophylaxis and mean FIX activity was maintained at 36.7%. Longer follow-up has reinforced that picture: in antibody-negative participants followed for four years, annualized bleeding rates remained low, endogenous FIX activity was around 39 IU/dL at year 4, and no participant had to return to continuous prophylaxis. These data explain why haemophilia B gene therapy was targeted in the webinar as being particularly promising in real life (Coppens et al., 2024; Raheja et al., 2025).
Going beyond severe haemophilia for prophylaxis
One of the most important changes in haemophilia management is that prophylaxis is no longer being considered from the lens of base line factor level. Recent reviews have indicated that people with non-severe haemophilia can still experience pain, decreased range of joint movement and impaired daily activity and quality of life and some may develop articular damage similar to that found in severe disease. ABR is 1.1 in median for all 104 people with non-severe haemophilia A including 1.6 in moderate disease (Dolan et al, 2024) – silent joint changes and depositions of hemosiderin even in joints presumed to be without bleeds (Dolan et al, 2024) This is the reason that the field is moving towards phenotype-based decisions. Expert opinion published in 2024 argues for some patients with non-severe haemophilia to have a bleeding phenotype which clearly justifies prophylaxis and a 2025 systematic review reported that more than 20% of patients with mild or moderate haemophilia A or B had a severe outcome such as joint bleeding, arthropathy, intracranial hemorrhage or inhibitor development. The practical message is simple – severity category still matters, but it should no longer be the only guide. Bleeding history, joint status, lifestyle and access to lower burden therapies are now as important (Pfrepper et al., 2024; Rodeghiero et al., 2025).
Existing clinical and access issues
Haemophilia care is advancing rapidly but the worst problems have not gone away. Inhibitor development is also one of the most serious complications particularly in haemophilia A, as this can make standard factor replacement less effective and complicates the planning of long-term treatment. Recent data from the EUHASS registry also indicate that modern treatment patterns are having an impact on the detection and interpretation of inhibitors: nowadays in 2022, 44% of severe HA patients before treatment are treated with emicizumab and the observed inhibitor incidence in this group had declined from 24% treatment years before 2016 to 6% in 2022. That is very encouraging, but it also means that delayed exposure to FVIII may complicate subsequent surveillance for the presence of inhibitors and comparison between products (Fischer et al., 2025). Access is an even larger problem. A 2025 global analysis estimated that the world prevalence of congenital haemophilia was 800,000 males, but most drug trials sites were in countries with high socioeconomic status compared to factor utilization being much lower in lower middle-income countries compared to high income countries. Reviews on equitable access make the same point in more direct terms: for most people with haemophilia globally, diagnosis and effective treatment remains out of reach, despite the continuing advancements in the therapies (Fedewa et al., 2025).
Conclusion
Real-world evidence is now at the core of haemophilia care in the 21st century. It demonstrates that prophylaxis is still at the centre, EHL products have made treatment less cumbersome, emicizumab has revolutionised haemophilia A treatment for many patients, and gene therapy has paved the way for the possibility of long-term factor independence in selected adults. At the same time these same real-life data remind us that no one treatment is the answer to everything: efficacy, durability, inhibitor risk, treatment burden, cost and access all matter when choosing a treatment. The trend in the field is to individualize prophylactic more and better use the digital support and also better focus on the outcomes that are important in everyday life (e.g., the health, freedom and participation of the joint). The true test of progress will be whether these advances will lead to improved care, not only for patients in expert centers, but also for patients everywhere.
References
Benemei, S., Mattia, C., & Dario, N. (2024). The good, the bad and the ugly of pain in haemophilia: Recent evidence on the epidemiology, molecular mechanisms and knowledge gaps preventing optimal treatment. Haemophilia, 30(3), 589–597. https://doi.org/10.1111/hae.15002
Blatný, J., Nielsen, E. M., Reitzel, S. B., McMillan, A. C., Danø, A., Bystrická, L., Kragh, N., & Klamroth, R. (2023). Real-world evidence on efmoroctocog alfa in patients with haemophilia A: A systematic literature review of treatment experience in Europe. Haemophilia : The Official Journal of the World Federation of Hemophilia, 29(4), 963–974. https://doi.org/10.1111/hae.14797
Buckner, T. W., Carpenter, S. L., Daoud, N., Kempton, C. L., Lee, L., Malec, L., McLean, T. W., Morton, P., O’Neill, C., Staber, J. M., Wang, M., Croteau, S. E., & Recht, M. (2025). Safety and Effectiveness of Emicizumab in People With Haemophilia A Enrolled in the ATHN 7 Haemophilia Natural History Study. Haemophilia. https://doi.org/10.1111/hae.70151
Carcao, M., Veena Selvaratnam, & Blatny, J. (2024). How much prophylaxis is enough in haemophilia? Haemophilia, 30(S3), 86–94. https://doi.org/10.1111/hae.14964
Chowdary, P., Carcao, M., Kenet, G., & Pipe, S. W. (2025). Haemophilia. The Lancet, 405(10480), 736–750. https://doi.org/10.1016/s0140-6736(24)02139-1
Chowdary, P., Khoo, L., Wang, M., Chambost, H., Chan, A. K. C., Willemze, A., & Oldenburg, J. (2025). Prospective, Observational Study of the Clinical Outcomes of FVIII Treatment in Adults and Adolescents with Severe Haemophilia A. TH Open, 09(CP). https://doi.org/10.1055/a-2621-9749
Christoph Bidlingmaier, Heller, C., Langer, F., Wolfgang Miesbach, Scholz, U., Oldenburg, J., Nüesch, E., Palmborg, H., Santagostino, E., & Tiede, A. (2024). Real-world usage and effectiveness of recombinant factor VIII/factor IX Fc in hemophilia A/B: final data from the 24-month, prospective, non-interventional PREVENT study in Germany. Research and Practice in Thrombosis and Haemostasis, 8(5), 102482–102482. https://doi.org/10.1016/j.rpth.2024.102482
Coppens, M., Pipe, S. W., Wolfgang Miesbach, Astermark, J., Recht, M., Paul, Ewenstein, B., Pinachyan, K., Galante, N., Sandra Le Quellec, Monahan, P. E., Leebeek, F. W. G., Giancarlo Castaman, Crary, S. E., Escobar, M., Gomez, E., Haley, K. M., Cedric, Kampmann, P., & Kazmi, R. (2024). Etranacogene dezaparvovec gene therapy for haemophilia B (HOPE-B): 24-month post-hoc efficacy and safety data from a single-arm, multicentre, phase 3 trial. the Lancet. Haematology. https://doi.org/10.1016/s2352-3026(24)00006-1
Dettoraki, A., Michalopoulou, A., Saslis, S., Dakou, K., Stamati, I., Thymianou, S., Kapsimali, Z., Papakonstantinou, O., & Pergantou, H. (2025). Real-World Use of Extended Half-Life Factor IX in Children with Haemophilia B. Life, 15(9), 1352. https://doi.org/10.3390/life15091352
Dolan, G., Fijnvandraat, K., Lenting, P. J., Catarino, C., & Lavin, M. (2024). Nonsevere Hemophilia: The Need for a Renewed Focus and Improved Outcomes. Seminars in Thrombosis and Hemostasis, 51(01), 058–067. https://doi.org/10.1055/s-0044-1786358
Fedewa, S. A., Valentino, L. A., Koo, A., Cafuir, L., Gillespie, T. W., Buckner, T. W., Tran, D. Q., Antun, A., & Kempton, C. L. (2025). Global patterns of hemophilia drug trials, hemophilia care, and health care measures. Research and Practice in Thrombosis and Haemostasis, 9(2), 102714. https://doi.org/10.1016/j.rpth.2025.102714
Fischer, K., Lassila, R., Peyvandi, F., Gatt, A., Gouw, S. C., Hollingsworth, R., Lambert, T., Kaczmarek, R., Carbonero, D., & Makris, M. (2025). Trends in Treatment of Severe Haemophilia and Impact on Inhibitor Assessment by the EUHASS Registry. Haemophilia. https://doi.org/10.1111/hae.70039
Gualtierotti, R., Solimeno, L. P., & Peyvandi, F. (2021). Hemophilic arthropathy: Current knowledge and future perspectives. Journal of Thrombosis and Haemostasis, 19(9), 2112–2121. https://doi.org/10.1111/jth.15444
Iorio, A., Königs, C., Reding, M. T., Rotellini, D., Skinner, M. W., Mancuso, M. E., & Berntorp, E. (2023). Prophylaxis use of clotting factor replacement products in people with non-severe haemophilia: A review of the literature. Haemophilia : The Official Journal of the World Federation of Hemophilia, 29(1), 33–44. https://doi.org/10.1111/hae.14676
Jiménez-Yuste, V. (2024). Non-factor Therapies for Hemophilia: Achievements and Perspectives. Seminars in Thrombosis and Hemostasis, 51(01), 023–027. https://doi.org/10.1055/s-0044-1796651
Kenet, G., & Fujii, T. (2024). Safety of recombinant activated factor VII for treatment of breakthrough bleeds in patients with congenital haemophilia A and inhibitors receiving emicizumab prophylaxis: Review of the real-world evidence. Haemophilia : The Official Journal of the World Federation of Hemophilia, 30(2), 267–275. https://doi.org/10.1111/hae.14933
Lewandowska, M., Nasr, S., & Shapiro, A. D. (2025). Emerging Therapies in Hemophilia: Improving Equitable Access to Care. Journal of Blood Medicine, Volume 16, 95–115. https://doi.org/10.2147/jbm.s490588
Mahlangu, J., Kaczmarek, R., von Drygalski, A., Shapiro, S., Chou, S.-C., Ozelo, M. C., Kenet, G., Peyvandi, F., Wang, M., Madan, B., Key, N. S., Laffan, M., Dunn, A. L., Mason, J., Quon, D. V., Symington, E., Leavitt, A. D., Oldenburg, J., Chambost, H., & Reding, M. T. (2023). Two-Year Outcomes of Valoctocogene Roxaparvovec Therapy for Hemophilia A. New England Journal of Medicine, 388(8), 694–705. https://doi.org/10.1056/nejmoa2211075
Olivieri, M., Yan, S., Yang, Y., Tomic, R., Linhoff, T., Zhang, X., Drelich, D., Jakobs, N., & Wolfgang Miesbach. (2025). Comparing Real-World Outcomes of Prophylaxis with Extended Half-life Factor IX (rIX-FP vs. rFIXFc and N9-GP) for Haemophilia B: An Analysis of Medical Chart Data from Germany. Advances in Therapy, 42(11), 5696–5707. https://doi.org/10.1007/s12325-025-03336-y
Pfrepper, C., Ettingshausen, C. E., Klamroth, R., Oldenburg, J., & Olivieri, M. (2024). Expert Opinion for Defining a Severe Bleeding Phenotype to Guide Prophylaxis in Patients with Nonsevere Hemophilia. Hamostaseologie, 10.1055/a2411–7416. https://doi.org/10.1055/a-2411-7416
Pratima Chowdary, Duran, B., Batty, P., Lowe, G., Jones, A., Pollard, D., Boyce, S., Motwani, J., Bahareh Amirloo, Musgrave, K., Hopper, D., Classey, S., Whitaker, S., Dunn, N., Bowyer, A., & Shapiro, S. (2024). UKHCDO gene therapy taskforce: Guidance for implementation of haemophilia gene therapy into routine clinical practice for adults. Haemophilia. https://doi.org/10.1111/hae.15125
Raheja, P., O’Connell, N., Verhamme, P., Kampmann, P., Lemons, R. S., Wang, F., Gill, S., Monahan, P. E., Le Quellec, S., & Leebeek, F. W. G. (2025). Etranacogene dezaparvovec in people with hemophilia B and without adeno-associated virus serotype 5 neutralizing antibodies: a 4-year subgroup analysis of the Health Outcomes with Padua Gene; Evaluation in Hemophilia B (HOPE-B) trial. Research and Practice in Thrombosis and Haemostasis, 10(1), 103321. https://doi.org/10.1016/j.rpth.2025.103321
Razmpoosh, E., Olasupo, O. O., Bhatt, M., Matino, D., & Iorio, A. (2025). Clotting factor concentrates for preventing bleeding and bleeding-related complications in previously untreated or minimally treated children with hemophilia A or B. Cochrane Database of Systematic Reviews, 2025(8). https://doi.org/10.1002/14651858.cd003429.pub5
Rezende, S. M., Neumann, I., Pantep Angchaisuksiri, Omolade Awodu, Boban, A., Cuker, A., Curtin, J. A., Fijnvandraat, K., Gouw, S. C., Gualtierotti, R., Makris, M., Nahuelhual, P., Niamh O’Connell, Saxena, R., Shima, M., Wu, R., & Rosendaal, F. R. (2024). International Society on Thrombosis and Haemostasis Clinical Practice Guideline for Treatment of Congenital Hemophilia A and B based on the GRADE methodology. Journal of Thrombosis and Haemostasis. https://doi.org/10.1016/j.jtha.2024.05.026
Rodeghiero, F., Ghiotto, L., Luca Pontalto, Casini, A., Giancarlo Castaman, Rezan Abdul‐Kadir, Berntorp, E., Imre Bodó, Manon Degenaar‐Dujardin, Fijnvandraat, K., Paolo Gresele, Key, N. S., Lassila, R., Frank, Lillicrap, D., Makris, M., Meijer, S., Mezzano, D., Noris, P., & Pabinger, I. (2025). Mild or moderate hemophilia is not always a mild or moderate bleeding disorder: Back to the clinical phenotype. HemaSphere, 9(3). https://doi.org/10.1002/hem3.70111
Samelson-Jones, B. J., Small, J. C., & George, L. A. (2024). Roctavian Gene Therapy for Hemophilia A. Blood Advances, 8(19). https://doi.org/10.1182/bloodadvances.2023011847
Srivastava, A., Santagostino, E., Dougall, A., Kitchen, S., Sutherland, M., Pipe, S. W., Carcao, M., Mahlangu, J., Ragni, M. V., Windyga, J., Llinás, A., Goddard, N. J., Mohan, R., Poonnoose, P. M., Feldman, B. M., Lewis, S. Z., Berg, H. M., & Pierce, G. F. (2020). WFH Guidelines for the Management of Hemophilia, 3rd Edition. Haemophilia, 26(s6).
van, Marloes de Kovel, Motwani, J., Chris van Geet, Nolan, B., Glosli, H., Carmen Escuriola Ettingshausen, Christoph Königs, Gili Kenet, & Fischer, K. (2024). Bleeding control improves after switching to emicizumab: Real‐world experience of 177 children in the PedNet registry. Haemophilia, 30(3), 685–692. https://doi.org/10.1111/hae.15015
von Drygalski, A., Chowdary, P., Kulkarni, R., Susen, S., Konkle, B. A., Oldenburg, J., Matino, D., Klamroth, R., Weyand, A. C., Jimenez-Yuste, V., Nogami, K., Poloskey, S., Winding, B., Willemze, A., & Knobe, K. (2023). Efanesoctocog Alfa Prophylaxis for Patients with Severe Hemophilia A. New England Journal of Medicine, 388(4), 310–318. https://doi.org/10.1056/nejmoa2209226
Wheeler, A. P., Abraham, A., Barnes, C., Brown Frandsen, R., d’Oiron, R., Eichler, H., Hampton, K., López‐Jaime, F. J., Lyu, C. J., Tavares, C. M. M., Nogami, K., Sutton, C., Windyga, J., Zulfikar, B., & Castaman, G. (2025). Real‐World Unmet Needs of Patients With Haemophilia A and Haemophilia B With or Without Inhibitors: End‐of‐Study Results From the explorer6 Non‐Interventional Study. Haemophilia, 31(5), 903–911. https://doi.org/10.1111/hae.70051
Yesim Dargaud, Leuci, A., Ruiz, A. R., & Sebastien Lacroix-Desmazes. (2024). Efanesoctocog alfa: the renaissance of Factor VIII replacement therapy. Haematologica. https://doi.org/10.3324/haematol.2023.284498
Young, G., Pipe, S. W., Kenet, G., Oldenburg, J., Safavi, M., Czirok, T., Nissen, F., & Mahlangu, J. (2024). Emicizumab is well tolerated and effective in people with congenital hemophilia A regardless of age, severity of disease, or inhibitor status: a scoping review. Research and Practice in Thrombosis and Haemostasis, 8(4), 102415. https://doi.org/10.1016/j.rpth.2024.102415
Zanon, E., Porreca, A., Napolitano, A., Simion, C., & Simioni, P. (2025). FVIII half-life products: A real-world experience. Thrombosis Research, 249, 109306. https://doi.org/10.1016/j.thromres.2025.109306
Zápotocká, E., Ovesná, P., Blažek, B., Čermáková, Z., Hajšmanová, Z., Hrdličková, R., Birke, P., Procházková, D., Smejkal, P., & Blatný, J. (2025). Emicizumab prophylaxis in current haemophilia A care in the Czech Republic—data from the Czech National Haemophilia Programme Registry. Research and Practice in Thrombosis and Haemostasis, 9(7), 103215. https://doi.org/10.1016/j.rpth.2025.103215

